
|
Professor of Anthropology |

|
Jonathan Marks |
|
DNA hybridization in the apes Technical issues |
|
Quite possibly the most widely cited work of primate evolution in the 1980s was that of Sibley and Ahlquist, using DNA to study primate phylogeny in papers published in 1984 and 1987 in The Journal of Molecular Evolution. They concluded that instead of yielding an effectively equidistant relationship among humans, chimpanzees, and gorillas, as most genetic data showed, their technique proved that humans and chimpanzees stood in a special phylogenetic relationship to one another, as closest relatives. The technique was called DNA hybridization, and they had been applying it extensively to problems of avian phylogeny already. But their data on the apes did not bear out their published phylogenetic conclusion. Serendipitously discovered by others (Vincent Sarich of the University of California at Berkeley, Carl Schmid of the University of California at Davis, and me), much of their data had been altered. These alterations had not been reported, and were thus unknown to reviewers and readers of their 1984 and 1987 papers. How important were they? Sibley et al. conceded in their 1990 paper that had it not been for their data alterations, "... it is virtually certain that Sibley and Ahlquist would have concluded that Homo, Pan and Gorilla form a trichotomy" [p. 225]. Thus, the only relevant questions are the precise nature of the previously unreported data alterations, and their validity. These can presumably be judged by most practicing scientists. |
|
The Unreported Sibley-Ahlquist "Corrections" |
|
Sibley and Ahlquist in their 1990 paper admit to three modes of unreported data alterations. The major "correction" they now admit to is as follows: in their words, it was performed by judging some data to be aberrant, "moving the aberrant point to the linear regression, determining the new [percent hybridization] value, and calculating the new T50H value" (p. 232). They plotted their experiments on Cartesian coordinates, one axis being how extensively the hybrids form (percent hybridization), and the other being the integrity of the hybrids (ΔT, melting temperature of perfectly bonded DNA strands of the same species minus the melting temperature of the hybrid DNA strands in this experiment; a low ΔT means the hybrid DNA is very similar to ordinary DNA, and thus well bonded, and thus derived from closely related species). |
|
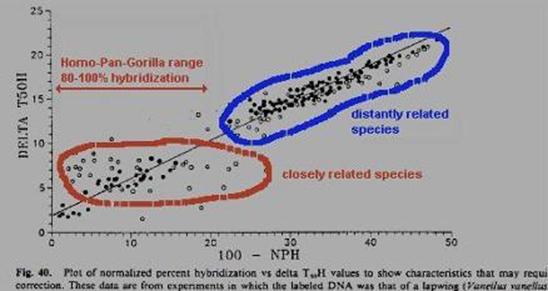
They found that human to chimp hybridizes very extensively, and the hybrids are well bonded; and that human to marmoset hybridizes poorly and those that form aren't so well bonded. They plotted their points, and found that they form a cloud that stretches from northwest to southeast. Then they asked their computer to generate the line which best describes the orientation of that cloud. That line is a linear regression, and that cloud is the difference between perfect determination of one variable by the other, and the real world of multiple causes for specific things. At the lower right, representing extensive hybridization and strong thermal stability of the hybrids (low ΔT), are the human-chimp and human-gorilla points. |
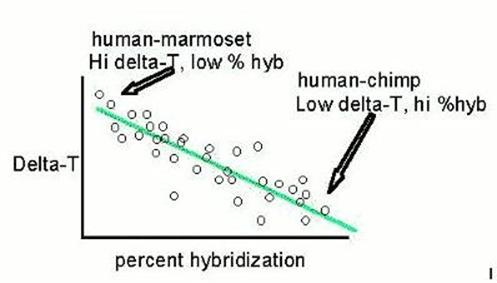
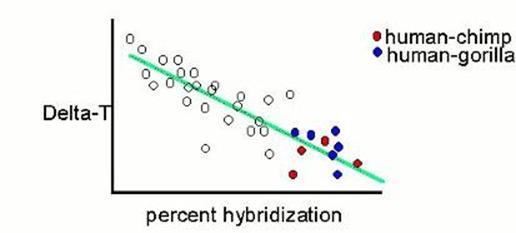
|
Then they looked at the human-gorilla points, and decided that they hybridized too much, and therefore have an artifactually deflated ΔT value. So they increased it a smidge. |
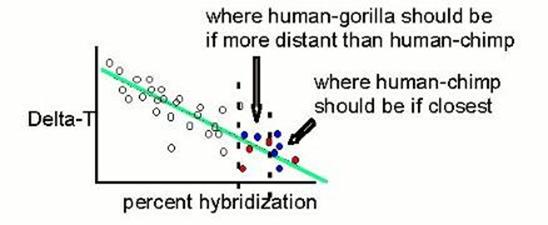
|
But they cannot possibly decide this unless they already know what the phylogeny is. |
|
In other words, they made this correction to get the "right" numbers on which to base their phylogeny, by imposing their phylogeny upon their data, and then altering the numbers to fit it. The T50H statistic incorporates the percent hybridization, which they have now altered. They give no criteria for judging specific points as "aberrant", and altered over 35% of their experimental results this way. As I noted in my "What's Old and New..." paper (American Journal of Physical Anthropology, 85:207, 1991) the experiments that indeed appear to have aberrant hybridization values are not the ones they consistently altered. |
|
Basically this is the best anyone can do to reconstruct their main data alteration procedure. They start off with human-chimp and chimp-gorilla points all mixed up (probably because they're equidistant). By knowing what phylogeny to expect, and imposing that on their data, they moved the points around accordingly, segregating the human-chimp from the human-gorilla. However, as I also noted in my "What's Old and New..." paper, Ahlquist in correspondence explained the published value of experiment 843-6 as due to this operation: "it just has a lower percent of hybridization than the others and can therefore be corrected". In their 1990 paper, however, they explain this value in Table 7 via alteration (3): "estimated correction for fragment length of driver DNA" (pp. 230-231). Consequently, I harbor deep suspicions that they did not actually even make the particular alterations they have now described. It should be noted that they treat their data as statistically independent in all of their published papers. |
|
Also, as they describe this regression line business in the 1990 paper, they illustrated it with bird data, rather than with the actual ape data that the paper is presumably about. And the bird data shows clearly that within the range of interest (high percent hybridization) there is no relationship between the two variables -- thermal stability and percent hybridization. |
|
So we know from their data that the DNA hybridization didn't work in discriminating closely related species. Why didn't it work? |
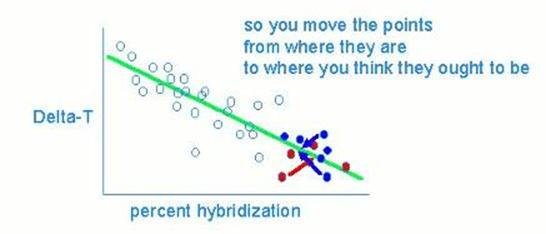
|
Paralogy |
|
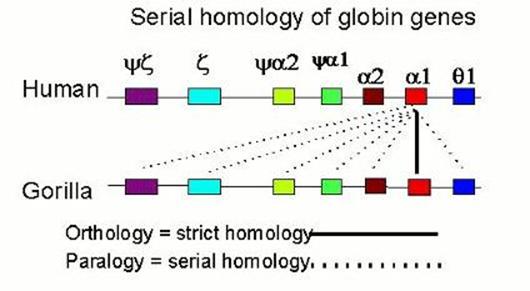
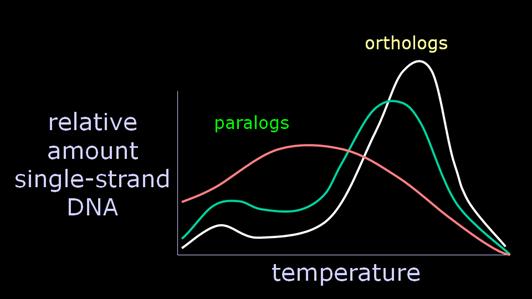
The problem is that there are other variables that the technique conflates with simple base-pair mismatch in measuring the T. One is the extent of hybridization, which is included in the T50H statistic of Sibley, but not in the Tm measurement of Powell. A second is more critical, and inheres in both measures -- the problem of serial homologs pairing in the hybrid DNA mixture. The technique was developed in the 1970s, when the conception of the genome generally tended to dichotomize "repetitive" against "unique-sequence" DNA (the latter presumably subsuming genes). This fails, however, to come to terms with the major discovery of the 1980s, the pervasive redundancy within all aspects of the genome. Not only are there repetitive elements distributed willy-nilly throughout the genome, but genes themselves appear invariably to cluster into families of related, duplicated genes. For example, the genes coding for the a-half of hemoglobin span about 30 kilobases and are all descended from a common ancestral sequence. Thus, if we wish to chop up and compare this DNA region of the human with that of the gorilla, as DNA hybridization does, each gene will in fact be fairly similar to no less than 7 genes in the other species. This relationship of molecular serial homology has come to be known as "paralogy", in contrast to proper homology or "orthology". |
|
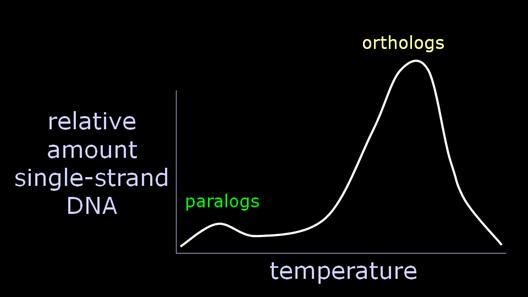
Now, let us say we are comparing closely related species, such as human and gorilla. Certainly the pairing of orthologs will be favored over paralogs, because the orthologs have only been differing since the taxa diverged, while the paralogs are have been differing since the genes duplicated. Further, we expect the minor component of paralogous pairing to be poorly bonded, and therefore to "melt" at a fairly low temperature. That is precisely what we find, when we examine the melting curves, which were at the heart of the original argument. |
|
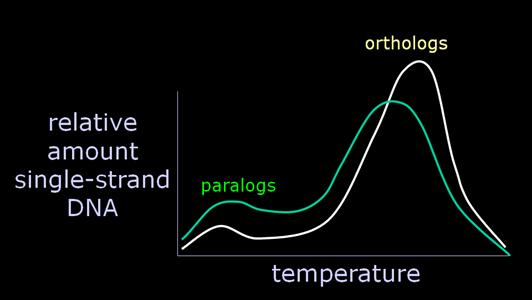
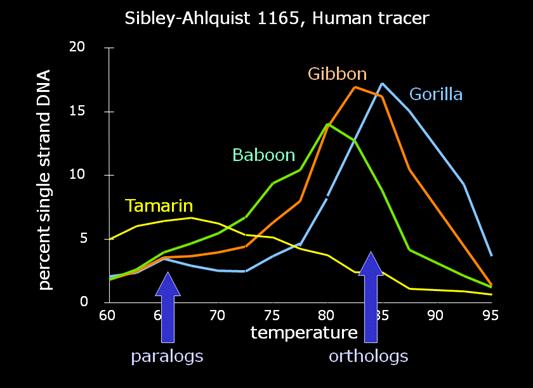
Essentially, a variable amount of hybrid DNA, from 0-15% of the total, will melt around 70 degrees, instead of around 84 degrees, with the orthologs. Recalling that the researchers regularly calculated and presented a median temperature, what we have is a significant uncontrolled variable which will strongly affect the calculation of the temperature at which 50% of the hybrid DNA is single-stranded. Consequently, Sarich began to argue strenuously for the calculation of the modal value as the "melting temperature" greatest significance. And we very specifically found in our 1988 J. Hum. Evol. paper that the modal value yielded no difference between human-chimp and chimp-gorilla. Quite simply, the variable amount of paralogous paired DNA is often considerably larger than the minuscule genetic differences Sibley was purporting to discriminate. But you can only determine this if you have the melting curves. |
|
That is why DNA hybridization did not, and does not, discriminate among very closely related species. The other side of the coin is also a problem. Let's say we want to study taxa which are distantly related. For the human-gorilla comparison, the orthologs have accumulated about 10 MY of divergence (i.e., the time the taxa diverged), while the paralogs have been diverging since the genes themselves duplicated, let's say an average of 100 MYA. Consequently, we can grossly estimate that the pairing of orthologs would be favored over the pairing of paralogs by a factor of about 10 to 1. |
|
Suppose we do a human-macaque experiment. Now the orthologs diverged 25 MYA, but the paralogs are still 100 MYA divergent. So the orthologs are now only favored by about 4 to 1. |
|
Further, since the orthologs are now more different from one another than the human-gorilla orthologs were, the main body of the melting curve is shifted to the left, and is blended with the satellite curve of the paralogs. It thus becomes more difficult to separate the information on orthologous DNA divergence (which one is presumably interested in, in studying the evolution of taxa) from paralogous DNA divergence (which is the evolution of gene families, interesting in itself, but not here). And if you do a human-lemur, the orthologs may only be favored by about 2 to 1 or less. |
|
And that is precisely what Sibley and Ahlquist found. In other words, this figure shows that DNA hybridization "works" -- for well-separated taxa. But for closely related taxa, the paralogous paired DNA introduces an error which undermines the ability to make very fine discriminations from orthologous DNA. And for very distantly related taxa, there is sufficiently little orthologous DNA in the mixture that it is simply hard to know what a "melting temperature" would actually be telling you. |
|
The Impossible 1989 "Replication" by Caccone and Powell |
|
The problem is that they (and others in this field) knew that showing the melting curves was the only way this problem could be identified, and so they refused to show them. Very significantly, the Caccone-Powell Evolution paper in 1989 also shows no melting curves. This paper claimed to have gotten the same numbers as Sibley and Ahlquist, in spite of using a different measurement, and in spite of not knowing what Sibley and Ahlquist's real numbers were! |
|
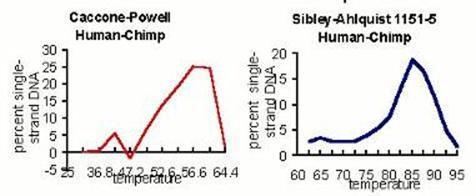
In fact, their data are not even as good as Sibley's. I was unsuccessful in acquiring any melting curves from them; Vince Sarich received three, none of which is as "clean" as the most comparable Sibley-Ahlquist experiments we have. The Caccone-Powell data have far larger secondary peaks, thus making the estimate of median melting temperature more crude; and how cruder data could possibly yield a more precise result is itself an interesting question. Their claims of having matched Sibley's numbers, thus vindicating them, and resolving the trichotomy, are remarkable. Why? The match is empirically false, as they did not match Sibley's real numbers, only the altered ones; and did not even match the Tms, only the altered T50H values. The vindication is a non-sequitur -- either Sibley's data resolve the trichotomy, or they do not and were altered to make them appear as if they did; nothing anyone else can do has any bearing on that. And whether or not Powell's data actually resolve the trichotomy would rest on a scrutiny of their own data and analysis. What, then, is the nature of the Caccone-Powell work? How could they have matched numbers that are (1) not real, and (2) not comparable, if they were real? The key appears to be a correction for fragment length applied to all DNA samples. Following sonication, nick translation, incubation, and S1 nuclease treatment, the DNA samples were run on a 2% agarose gel, and lengths estimated for the DNA samples. These are given to the nearest single nucleotide in the 1989 paper, a remarkable degree of precision after this treatment. |
|
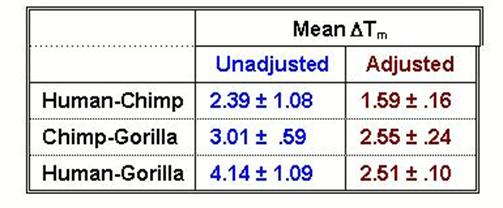
Calculating from the appendix to the paper, it can be easily shown that prior to this correction, the data neither match the published Sibley numbers, nor resolve the trichotomy. Subsequent to the correction, they do both. The effects of the correction can be seen in two ways from the numbers given in their paper's Appendix. First, in one set of Homo-Pan comparisons (HU2-PP2), the unadjusted melting temperature (tm) is given as 51.85 (standard error of 0.20). In another (HU2-PT2), the unadjusted melting temperature is given as 51.30 (standard error 0.04), yielding a difference between the two sets of experiments comparing equidistant taxa of over half a degree. After correction, the melting temperatures (Tm) are given as 56.90 and 56.87, differing now by only 0.03 degrees. The mean lengths of these two sets of DNA samples are given as 99 and 90, respectively, with no associated scatter. This difference of precisely 9 nucleotides for two samples of whole genomic DNA following sonication, nick translation, hybridization, and enzymatic digestion, is very likely well beyond the limits of resolution or of biochemical significance to DNA samples treated in this manner, and is consequently very counterintuitive. Nevertheless, the adjustment based on this measurement brings these two experiments into very close harmony. Second, numbers given in that Appendix before and after numerical correction can be averaged to reveal the effect of the curious length adjustment on the final values. |
|
The precise length-correction adjustment has the effect of reducing the standard deviation by up to an order of magnitude, of harmonizing the chimp-gorilla and human-gorilla numbers, and of bringing the three sets of value into harmony with the published Sibley numbers. Without the length correction, there is no resolution, and there is no match to the published Sibley numbers, which are the two central claims of the Caccone-Powell paper. Further, the purported match between the published Sibley-Ahlquist numbers and the published Caccone-Powell numbers is itself exceedingly misleading. Sibley-Ahlquist measured T50H, the median melting temperature of the DNA which could have formed hybrids; Caccone-Powell measured Tm, the median melting temperature of the DNA which actually formed hybrids. The difference is the incorporation of the extent of hybridization into the calculation of T50H. Until the 1990 Sibley et al. paper, the published Sibley-Ahlquist numbers were given only as T50Hs. In the 1990 paper, they gave the unaltered Tm values for their experiments as well. These do not resolve the trichotomy, but more importantly, they do not match the Caccone-Powell numbers, to which they are more comparable. |
|
Thus, the match to the Sibley numbers is completely false, the assertions on its behalf are highly misleading, and the resolution of the trichotomy is dubious. There are many possible explanations for this unfortunate situation. The simplest explanation is that the fragment-length numbers, whose precision can have very little biochemical significance, were manipulated to generate the results; and the explicit association of this work to the Sibley-Ahlquist work was intended to mislead the scientific community about the quality and integrity of the latter. The ultimate problem for science is that if DNA hybridization works at all, it is only for a very narrow range of phylogenetic questions. |

|
Conclusions |
|
The best-case scenario for the Sibley work is that the analysis itself was done naively, the omission of the vital information was coincidental; and the refusal of the authors to allow others to see their data was also coincidental. In the worst case, they knew that their data analysis was illegitimate; the omission was required to conceal the illegitimate analysis; and the sequestering of the documentation was required to conceal the omission of the data alteration procedures from the published papers. My opinion is that the latter is the most parsimonious explanation for (1) the consistent omission of the key analytic procedures from their papers; (2) the reluctance to provide interested colleagues with the data for alternative analyses; and (3) the invocation of conclusions from other studies to justify the analysis of their own data. To the extent that the Powell work ties itself to the Sibley work, it must also be examined with a great deal of caution and skepticism.
|
|
The most unfortunate aspect of this story is that the research is intimately associated with the biology department at Yale, and despite extensive publicity and discussion in the primary literature, it was never the subject of any investigation or adjudication. It is quite likely that if such an investigation had been undertaken, it would have concluded that the scientific community had been egregiously and willfully misled by these papers, and that this has been an example of the classic triad of fraud, cover-up, and retaliation. |

|
Jonathan Marks |
|
email: jmarks@uncc.edu |